Abstract
L’Association for European Cardiovascular Pathology (AECVP) ha recentemente pubblicato su Virchows Archiv un documento sulla diagnosi autoptica delle Cardiomiopatie ereditarie. La revisione dei passaggi salienti dell’articolo consente di compendiare i principali segni patognomonici o suggestivi di Cardiomiopatia ereditaria, elicitando spunti di riflessione su operatività medico-legali e normativa privacy.
Se vuoi leggere l’intera review, clicca qui – Sheppard MN et al.; Association for European Cardiovascular Pathology (AECVP). Genetically determined cardiomyopathies at autopsy: the pivotal role of the pathologist in establishing the diagnosis and guiding family screening. Virchows Arch. 2023 Apr;482(4):653-669. doi: 10.1007/s00428-023-03523-8
. . .
Le Cardiomiopatie (CMP) sono entità nosografiche che comprendono numerose patologie, distinte per presentazione clinica, meccanismo patologico e substrato eziologico, ma accomunate dall’interessamento primario del miocardio. In particolare l’eziopatogenesi può essere su base genetica o acquisita.
In particolare, l’American Heart Association (AHA) ricomprende nel gruppo delle CMP le patologie del miocardio, associate a disfunzione elettrica o meccanica, che frequentemente comportano ipertrofia o dilatazione ventricolare.
Per converso, la European Society of Cardiology (ESC) definisce la CMP una patologia del miocardio nella quale il muscolo cardiaco è alterato dal punto di vista strutturale e funzionale, in assenza di coronaropatie, ipertensione, patologia valvolare, alterazioni congenite. La Classificazione ESC compendia 4 macrogruppi di CMP, sulla base delle peculiari alterazioni morfologiche e funzionali: Cardiomiopatie Dilatative (DCM), Cardiomiopatie Ipertrofiche (HCM), Cardiomiopatie Restrittive (RCM), Cardiomiopatie Aritmogene (ACM).
Cardiomiopatie Ereditarie: Segni suggestivi e diagnosi differenziale
Cardiomiopatie Ipertrofiche (HCM)
Il riscontro di ipertrofia ventricolare sinistra (spessore miocardico >1.5 cm, escluse trabecolature) in assenza di cause sottese – ad es. stenosi aortica o ipertensione – deve ritenersi suggestivo per HCM. La HCM è infatti una della cardiomiopatie ereditarie più diffuse: un soggetto su 500 ne è affetto. Il substrato eziologico è rappresentato da alterazioni autosomiche dominanti di geni che codificano per proteine del sarcomero miocardico.
Come anticipato, l’aspetto macroscopico del cuore nei casi di HCM ereditaria è caratterizzato da aumento non uniforme dello spessore del miocardio ventricolare sinistro e del setto interventricolare, con risultante potenziale aumento dei diametri cardiaci e del peso. Il volume della cavità ventricolare sinistra è usualmente ridotto, con possibile ispessimento e degenerazione balloniforme del lembo anteriore della mitrale per frizione. Vi è inoltre aumentata incidenza di bridging miocardico.
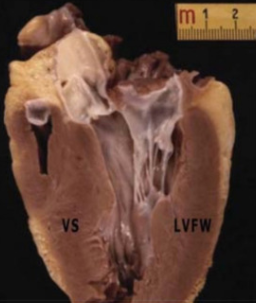
Immagine tratta da Vio R, et al: Hypertrophic Cardiomyopathy and Primary Restrictive Cardiomyopathy: Similarities, Differences and Phenocopies. J Clin Med. 2021 May 1;10(9):1954. doi: 10.3390/jcm10091954.
A livello microscopico, invece, si rileva ipertrofia dei miocardiociti – con forme bizzarre, pleomorfismo nucleare, ipercromasia – e disarray. Altra caratteristica saliente è la fibrosi interstiziale, nel cui contesto spesso insistono vasi arteriosi con ispessimento concentrico delle tonache intima e media.
Tuttavia alcune patologie ereditarie sistemiche possono presentare quadri sovrapponibili a quanto descritto: le patologie da accumulo di glicogeno, la malattia di Fabry e alcune patologie mitocrondriali, ad esempio, differiscono per la vacuolizzazione dei miociti e/o flogosi, mentre le desminopatie solo raramente interessano primariamente il cuore. In questi casi solo lo studio ultrastrutturale può chiarire la diagnosi.
Cardiomiopatie Aritmogene (ACM)
Storicamente nota come Displasia Aritmogena del Ventricolo Destro, è stata successivamente inquadrata con l’attuale designazione nosografica in quanto termine più idoneo a descrivere un processo di progressiva (dall’epicardio all’endocardio)sostituzione del miocardio con tessuto cicatriziale fibroadiposo ventricolare, con possibili varianti ad espressione predominante a livello del ventricolo sinistro.
Tuttavia, mentre la ACM nel ventricolo destro è prevalentemente ereditaria, aree cicatriziali a livello del ventricolo sinistro possono essere espressione di pregressi eventi ischemici o flogistici.
La prevalenza della ACM è di 1:2000-5000 ed è una causa ben nota di morte improvvisa giovanile o durante attività sportiva. La base è una mutazione dei geni codificanti per proteine delle giunzioni intercellulari.
A livello macroscopico, il cuore affetto da ACM si presenta con diametri e peso regolari o lievemente aumentati, mentre lo spessore miocardico è sovente, ma non sempre, ridotto. Alla sezione sarà possibile riconoscere focali aree discromiche giallastre o biancastre ad esordio epicardico. La distribuzione focale delle lesioni rende necessario effettuare plurime sezioni trasversali biventricolari.
I segni distinitvi dell’alterazione, tuttavia, sono riscontrabili solo a livello microscopico, con evidente fibrosi interstiziale e sostitutiva, alternata a tessuto adiposo specie a livello del ventricolo destro. In particolare è significativo il riscontro di aree di sostituzione adiposa a livello del cono di efflusso del ventricolo destro.
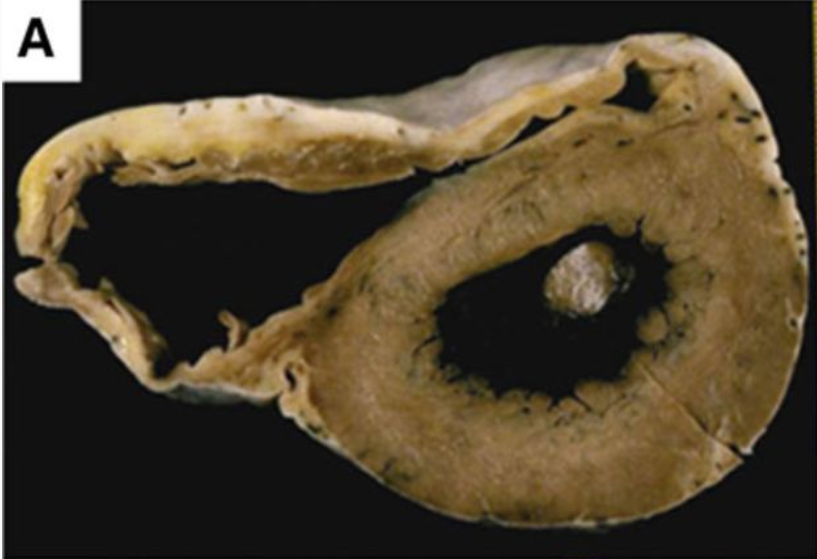
Immagine tratta da Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017 Sep 15;121(7):784-802. doi: 10.1161/CIRCRESAHA.117.309345.
La diagnosi differenziale prevede l’esclusione di pregressi eventi ischemici o miocarditi, sovraccarico del ventricolo destro per alterazioni anatomiche congenite, effetti cronici di sostanze esogene, distrofie muscolari, sarcoidosi cardiaca isolata, malattia di Chagas.
Cardiomiopatie Dilatative (DCM)
È la più comune forma di cardiomiopatia, con una prevalenza di 1:250. Il 30-50% dei casi è su base genetica. Il riscontro di cardiomegalia e dilatazione della cavità ventricolare sinistra, in assenza di patologia coronarica, valvolare o ipertensione, deve orientare per forme ereditarie. Sono infatti oltre 50 i geni coinvolti nei casi di DCM familiare, codificanti per proteine del citoscheletro, del sarcolemma, rivestimento nucleare, canali ionici e giunzioni intracellulari. La causa più comunemente identificata è l’alterazione della titina.
È stato osservato come, anche in casi con chiara presenza di fattori di rischio per DCM acquisita (abuso di alcol, cocaina, gravidanza, miocarditi, emocromatosi, sarcoidosi, terapia con antracicline), vi fosse ricorrenza di una predisposizione ereditaria nel 20% dei casi. Tanto indicherebbe un più ampio ricorso ad approfondimenti genetici.
La valutazione macroscopica del cuore nei casi di DCM offre a considerare aumentato diametro trasverso, cavità ventricolare sinistra dilatata, aumento delle circonferenze degli annulus mitralico e tricuspidale. Alla sezione ventricolare si potranno osservare aree di fibrosi, trombi intramurali da ipocinesi, ipertrabecolazione. Al microscopio il quadro potrà essere rappresentato da aumento del diametro dei miociti, alterazioni morfologiche del nucleo con discromie perinucleari, firbrosi interstiziale o sostitutiva.
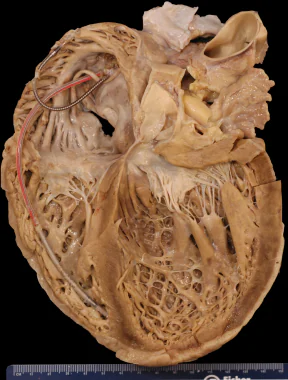
Immagine tratta da Dilated Cardiomyopathy Pathology, Updated: Nov 04, 2015. Author: Allen Patrick Burke, MD; Chief Editor: Allen Patrick Burke, MD su MEDSCAPE.
Cardiomiopatie Restrittive (RCM)
La RCM è entità nosografica ancora poco conosciuta, a scarsa prevalenza e solo raramente causa di morte improvvisa. È caratterizzata da ventricoli non dilatati e non ipertrofici, a fronte di una evidente dilatazione degli atri.
Le più comuni alterazioni genetiche sottese a RCM primarie includono mutazioni di geni codificanti la catena pesante della beta miosina e la troponina I. Tra le cause alternative si annoverano patologie infiltrative e malattie da accumulo, malattie mitocondriali, malattie sistemiche come l’emocromatosi.
Il quadro anatomopatologico è caratterizzato da dilatazioni atriali con possibili trombosi intramurali e diffusa fibrosi interstiziale.
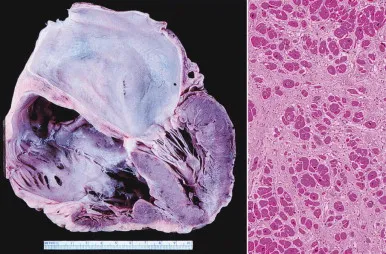
Immagine tratta da thoracickey.com.
Protocolli Operativi
Un riscontro diagnostico o autopsia giudiziaria dovrebbe essere effettuato in tutti i casi di Morte Improvvisa. Sul punto occorre precisare che la strutturazione dell’articolo 37 del DPR 285/1990, così come modificato dalla Legge 24/2017, con riconoscimento (comma 2 bis) ai familiari di un loro ruolo in merito all’esecuzione di riscontro diagnostico, non sovverte la poziorità del sanitario richiedente l’accertamento nei casi di opposizione da parte di uno o più familiari.
Tuttavia, quale corollario al comma 2 bis della citata norma, è estensibile un richiamo al diritto dei familiari ad essere preventivamente informati delle finalità e – successivamente – delle risultanze dell’autopsia effettuata nei casi di morte improvvisa.
Ed è dunque in questi due distinti momenti comunicativi (richiesta dell’esecuzione del riscontro diagnostico da parte dei familiari ovvero loro richiesta degli esiti del riscontro stesso) l’occasione ideale per poter acquisire dalla famiglia informazioni utili ad un migliore inquadramento dal punto di vista clinico-anamnestico. Nell’ambito delle cardiomiopatie su base genetica, in particolare, rilievo preminente è assunto dall’anamnesi familiare, con individuazione di ulteriori casi di morte improvvisa o morte per causa cardiaca nell’albero genealogico.
Ecco allora, è di piana evidenza come in questi casi l’autopsia debba essere necessariamente completa e corredata di tutti gli ancillari approfondimenti, in primis quello tossicologico. Escluse le cause extracardiache di morte improvvisa, dunque, l’analisi macroscopica dell’organo cardiaco dovrà essere effettuata secondo le Guidelines for autopsy investigation of sudden cardiac death dell’AECVP (LINK), con accurato rilievo e documentazione, anche fotografica, dei segni salienti.
Prelievi Autoptici
Quanto alla campionatura per l’analisi istopatologica, è raccomandata l’esecuzione di una completa mappatura di una sezione biventricolare cardiaca come da immagine.
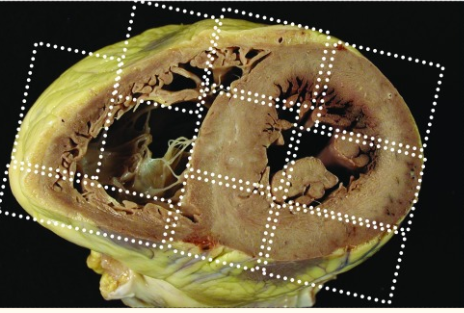
Immagine tratta da Basso C, et al. Association for European Cardiovascular Pathology. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch. 2017 Dec;471(6):691-705. doi: 10.1007/s00428-017-2221-0
Nei casi dubbi l’organo in toto sarà idoneamente preservato in soluzione formolica al fine di poter ottenere eventuali second opinion da centri specializzati in Patologia Cardiovascolare.
È raccomandabile inoltre il prelievo di sangue periferico e tessuto splenico per le successive indagini genetiche nei casi di cardiomiopatie ereditarie.
Cardiomiopatie e Indicazioni al Counseling Genetico
Ma in quali casi è opportuno indirizzare i familiari del deceduto ad un Counseling Genetico?
Soccorrono, a fugare eventuali dubbi, il citato documento del 2017 dell’AECVP e le Raccomandazioni Europee del 2019 della Società Europea di Genetica Umana (ESHG) alle quali si rimanda per i riferimenti bibliografici.
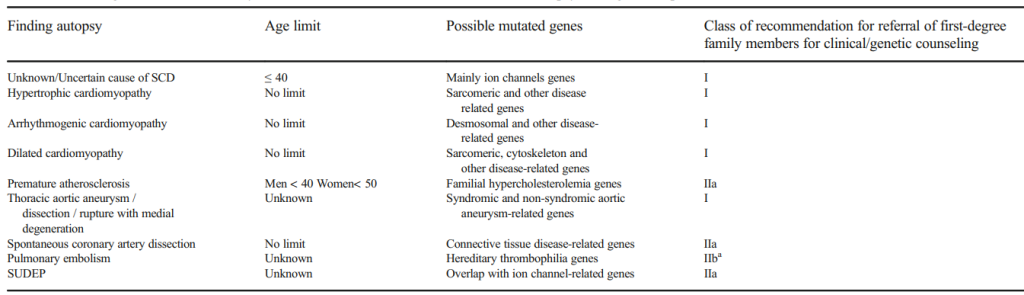
Tabella tratta da Basso C, et al. Association for European Cardiovascular Pathology. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch. 2017 Dec;471(6):691-705. doi: 10.1007/s00428-017-2221-0.
La tabella riportata indica, per ogni rilievo cardiopatologico osservato in autopsia, un limite di età del deceduto entro il quale è indicato avviare i familiari ad approfondimenti clinici ed eventualmente genetici, con una indicazione del rango di evidenze dal quale discende la raccomandazione (Classe I Raccomandato, Classe IIa Utile, Classe IIb Da Considerare).
Le raccomandazioni della ESHG del 2017, invece, integrano la precedente tabella con una flowchart per l’affidamento della famiglia del deceduto ad un team multidisciplinare:
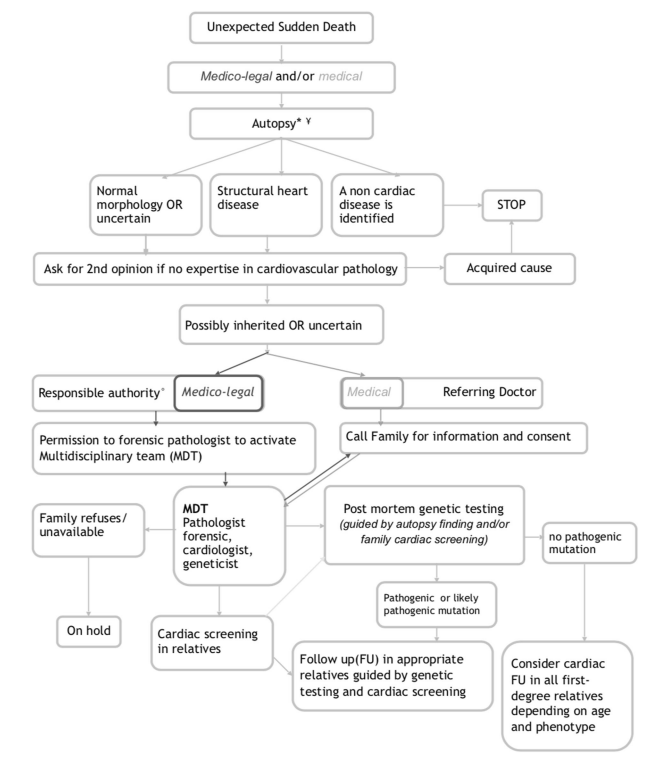
Sul punto si rammenta che il Counseling Genetico è annoverato tra i LEA di cui al DPCM 12 gennaio 2017 (ALLEGATO 4) e pertanto gratuito.
Diritto a (Non) Sapere
L’articolo 2 del Dlgs 196/2003 e successive modificazioni garantiscono l’accesso ai dati sulla salute del defunto a soggetti portatori di un interesse proprio, meritevole di protezione, quale la Salute. Inoltre, l’Autorizzazione n. 8/2016 – Autorizzazione generale al trattamento dei dati genetici – all’art. 3 punto 1.b stabilisce che possono essere trattati dati genetici ed utilizzati campioni biologici per finalità di “tutela della salute, con particolare riferimento alle patologie di natura genetica […]”.
Nel caso in cui l’interessato sia deceduto “il trattamento può comprendere anche dati genetici estrapolati dall’analisi dei campioni biologici della persona deceduta, sempre che sia indispensabile per consentire al terzo di compiere una scelta riproduttiva consapevole o sia giustificato dalla necessità, per il terzo, di interventi di natura preventiva o terapeutica”.
Se quindi la vigente normativa prevede la possibilità di utilizzo di campioni genetici per finalità di tutela della salute, nondimeno gli accertamenti autoptici e le successive indagini molecolari possono elicitare problematiche dal punto di vista bioetico.
È un fatto che informare i familiari sulle cause del decesso esorbiti gli obiettivi primari di una Autopsia Giudiziaria. Parimenti, il Riscontro Diagnostico potrebbe essere richiesto dal medico curante o dai familiari per ragioni diverse dall’identificazione di patologie a carattere familiare o genetico (per es. mera volontà di conoscere quale sia stato l’evento esiziale o, non di rado, finalità assicurative).
È parimenti un fatto che, nei casi di cardiomiopatie su base genetica, ma non solo, i parenti del defunto potrebbero essere inconsci portatori di un rischio aumentato di morte improvvisa e che tale consapevolezza potrebbe da un lato consentire l’accesso a cure e terapie potenzialmente efficaci ma dall’altro, specie in caso di patologie inemendabili e non sanabili, avere ingenti ripercussioni sulla serenità e qualità della vita del familiare.
Si configura dunque un contrasto tra l’imperativo etico di mettere in guardia e il ‘diritto a non sapere’. Contrasto, occorre dirlo, di non semplice risoluzione ed oggetto di ampio ed attuale dibattito nella Comunità Scientifica internazionale (LINK).
Nelle more di una auspicabile indicazione legislativa chiara, una parziale soluzione al dilemma può consistere nella richiesta ai familiari coinvolti nel riscontro diagnostico di determinarsi in anticipo – vale a dire prima di ricevere gli esiti dell’accertamento autoptico – rispetto a tale eventualità.
In altri termini, è proponibile la previsione di uno specifico consenso alla comunicazione di potenziali diagnosi inattese relative a patologie di certa o probabile genetica, da raccogliersi su apposita modulistica (all’atto del colloquio prodromico all’autopsia ovvero al momento della protocollazione della richiesta motivata di acquisizione degli esiti del riscontro diagnostico) in modo da rispettare a pieno la volontà dell’avente diritto.
VUOI APPROFONDIRE QUESTO ARGOMENTO?
Vedi anche: Dati relativi alla salute del soggetto defunto: chi ha diritto di accesso? e Infiammazione miocardica e Covid: causa di morte o reperto incidentale?






